長らく新薬業界とジェネリック業界の利害調整に手間取っていた新薬データ保護制度(https://www.kawamotobbp.jp/articles/2560)ですが、今回、中国政府は新薬「データ保護」規則(案)(药品试验数据保护实施办法(征求意见稿):https://www.nmpa.gov.cn/xxgk/zhqyj/zhqyjyp/20250319181537196.html)を公表しました。新薬について、
- 新規有効成分を含有する新薬 ⇒ 6年
- 新規適応症・新製剤・新投与ルート等 ⇒ 3年
のデータ保護を付与し、その間、原則、ジェネリック薬は承認しない、としました。
但し、新薬に関する特許期間の延長制度(https://www.kawamotobbp.jp/articles/2375)と同様に、新薬のデータ保護を受ける為には、中国での早期開発等の条件が付いています。
まだ、(案)の段階ですが、かなり煮詰まって来ており、今後、最終化の上、年内には施行予定とされています。
1.新薬「データ保護」規則(案)の全容
「データ保護」とは
新規有効成分(低分子医薬、バイオ医薬、ワクチン)を含有する「創新薬」と既存薬の新規適応症・新製剤・新規投与ルート等の「改良型新薬」について、医薬品企業(先発企業)がNMPA(中国医薬品管理局)に提出した上市承認申請に添付した有効性・安全性・CMC等のデータに対して保護が与えられます。「創新薬」は6年、「改良型新薬」は3年のデータ保護期間(規則案§5,6)が付与され、この保護期間中は、原則、ジェネリック薬の上市承認は下りません。
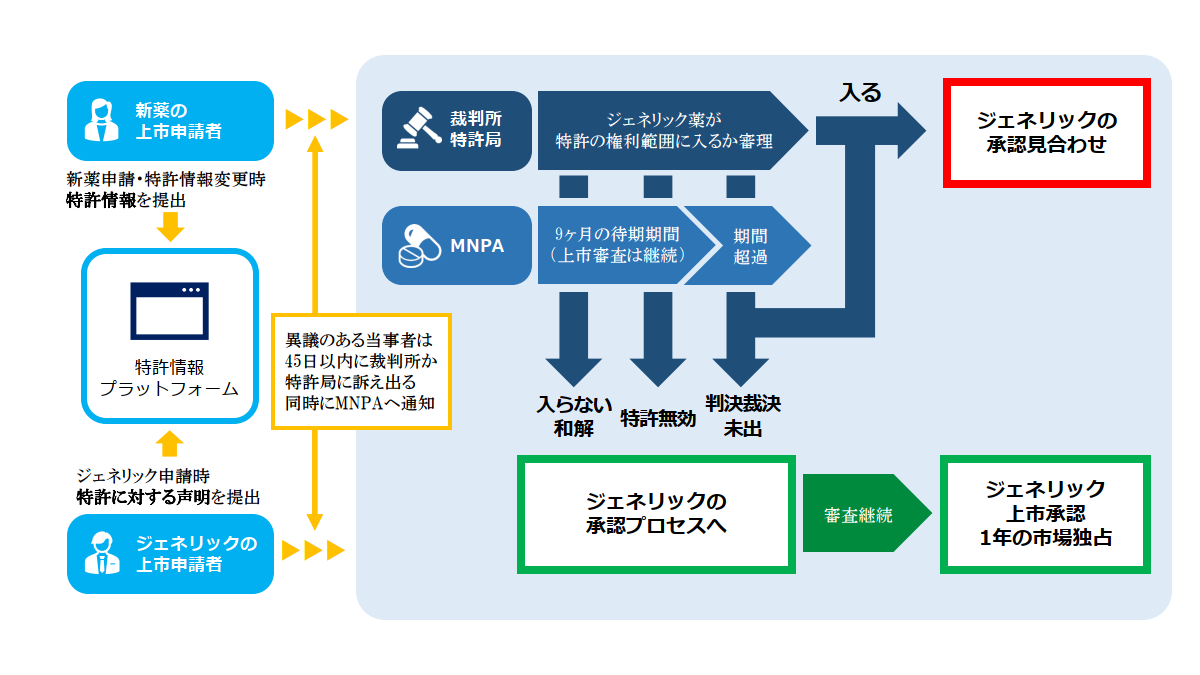
ただし、データ保護期間満了後であっても、当該「創新薬」・「改良型新薬」が先発企業の特許でカバーされていた場合には、ジェネリック薬の上市承認申請は、特許侵害紛争のリスクを抱えます。特許でカバーされている場合には、ジェネリック薬の審査段階で、patent linkageの制度(https://www.kawamotobbp.jp/articles/1615)の下で、侵害の紛争処理が行われます。
日本では、再審査期間中(新規有効成分:8年)は、ジェネリック薬に対して原則、販売承認が付与されません。中国の「データ保護」は、日本のこの「再審査制度」に相当する制度です。
日本の再審査制度との違い
日本の再審査制度にはない中国の「データ保護制度」の特徴は、下記の二点です。
- 「創新薬」・「改良型新薬」の中国での上市承認申請時以前に、海外(日本を含む)で対応の医薬品の販売承認が下りていない事。その場合には、夫々、前記の6年、3年の全期間のデータ保護が付与されます。
他方、中国での上市承認申請時に海外(日本を含む)で既に販売承認が下りていた場合、保護期間は、中国での上市承認申請が遅れた年月に相当する期間が短縮される。 - 海外で既に販売承認が下りているオリジナル新薬を、中国企業等の第三者が中国で確認試験(小規模Ph III)を実施し、当該オリジナル新薬のジェネリック薬を申請し、販売承認が下りた場合、3年間のデータ保護期間が付与される。
「創新薬」と「改良型新薬」とは
①「創新薬」(低分子、バイオ医薬、ワクチン):新規有効成分(NCEを含む)であって中国の国内外で未上市の薬剤。
詳しくは、海外(日本を含む)での最初の販売「承認日」の前に中国で上市承認「申請」をすれば、中国の医薬品登録分類上、「創新薬」として、「1類」に分類される。
尚、中国での開発が遅れて、海外(日本を含む)での最初の販売承認日以降に中国で上市承認申請をする場合、「5.1類」に分類される。海外上市後の中国での申請であることから、NMPAに提出が必要なデータの範囲は、「1類」よりも狭い範囲で良く、中国で確認臨床試験(小規模Ph III)を実施すればよい。
②「改良型新薬」(低分子医薬):中国の国内外で未上市の薬剤であって、既知の有効成分を改良した薬剤。下記を含む。
- 承認済の当該有効成分に対する、新規の光学異性体・エステル・塩等
- 当該有効成分を含有する承認済み製剤に対して、新規の適応症・新製剤・新投与ルート等
尚、バイオ医薬、ワクチンについても上記に準じる。
詳しくは、海外(日本を含む)での最初の販売「承認日」の前に中国で対応の薬剤の上市承認「申請」をすれば、中国の医薬品登録分類上、「改良型新薬」として、「2類」に分類される。
尚、中国での開発が遅れて、海外(日本を含む)での最初の販売承認日以降に中国で上市承認申請をする場合、「5.1類」に分類される。「5.1類」は、海外での上市後の中国申請であることから、「2類」に比べ、中国で実施すべき臨床試験は小規模で済む。
例外(規則案§3)
「データ保護」期間中であっても、ジェネリック企業が開発費を投入して臨床試験を実施し、先発企業の当該保護されたデータと同レベルのデータを取得の上、上市承認申請した場合には、要件を満たせば、承認されます。尚、先発企業が特許を有する場合は、前記の通り、特許侵害のリスクにさらされます。
また、保護対象となっているデータを所有する「創新薬」・「改良型新薬」の上市承認の取得者(先発企業)から同意があれば、ジェネリック薬は同様に承認されます。
ジェネリック薬の上市承認の申請のタイミングの制限(規則案§11)
ジェネリック薬は、「データ保護」期間中であっても、BE試験(生物学的同等性試験)等の臨床試験を実施して、「データ保護」期間の満了前1年以降、ジェネリック申請が可能です。NMPAは、当該ジェネリック薬の審査を行い、要件を満たせば、「データ保護」期間の満了を待って、上市承認を付与する。
2.「創新薬」「改良型新薬」のデータ保護期間
原則(規則案§5,6)
| カッコ内は薬事法上の登録分類 | 保護期間 |
| 創新薬 (1類) | 中国での創新薬の上市承認の付与日から「6年」 |
| 改良型新薬 (2類) | 中国での改良型新薬の上市承認の付与日から「3年」 |
中国での申請が海外の販売承認付与よりも遅い場合(規則案§5,6)
N期間(遅れた年月)=(中国での上市承認申請日)―(海外での最初の販売承認付与日)
| カッコ内は薬事法上の登録分類 | 保護期間 |
| 創新薬 (5.1類等) | 「6年」- N期間 |
| 改良型新薬 (5.1類等) | 「3年」- N期間 |
<例1>中国開発が遅れた場合
(前提)
- 「創新薬A」について日本での販売承認日:2025年8/1
- 中国での上市承認申請日:2026年8/1
- 中国での上市承認付与日:2027年8/1
(データ保護期間)
- N期間(遅れた年月)=1年
- 保護期間=6年-1年
=5年 - 従って、保護期間の満了日=中国での上市承認付与日から5年
=2032年8月1日 - 尚、ジェネリック薬は、保護期間の満了一年前以降、即ち、2031年8月1日以降に上市承認申請が出来、要件を満たせば、保護期間満了後に上市承認が下りる。
尚、上記の「創新薬A」が改良型新薬の場合、データ保護期間は3年であることから、上記の前提のように遅れた年月が1年の場合、保護期間は、「2年」(3年-1年=2年)となる。
新適応症の「データ保護」期間
「創新薬」の最初の上市承認取得以降、追加の臨床試験を実施して取得したデータに基づく、新規の適応症の上市承認申請については、承認後、その都度(新適応症毎)、当該追加データについて、データ保護が付与される。尚、新規の適応症は、「改良型新薬」(2類)に分類されることから、データ保護期間は、当該新適応症の上市承認後、3年である。
<例2>最初の上市承認の保護期間 vs 新規の適応症の保護期間
(前提)
- 中国での適応症Xの「創新薬」としての最初の上市承認:2027年8月1日
- 上記創新薬に対する、中国での新適応症Yの追加の新適応症の上市承認日:2028年8月1日
(データ保護期間)
- 適応症X:保護期間=6年
データ保護満了日=2033年8月1日 - 新適応症Y:保護期間=3年
データ保護期間満了日=2031年8月1日
(問題点)
データ保護期間が先に満了する新適応症Yについて、データ保護期間が満了(2031年8/1)後、ジェネリック薬が直ぐに承認され、ジェネリック薬(新適応症Y)が市場に出た(創新薬に特許がない前提)場合、データ保護期間が満了していない適応症Xの用途にジェネリック薬が現場で転用される可能性あり。従って、先発企業は、適応症XとYで製剤型を変更する等の工夫が必要となって来る。尚、それが困難な場合には、市場規模の大きい適応症を重視した申請戦略を取る必要がある。
<例3>中国開発が遅れた場合(日本、中国が同一適応症)
(前提)
- 「創新薬A」の「適応症X」について日本での販売承認日:2025年8月1日
- 中国での「適応症X」の上市承認申請日:2026年8月1日
- 中国での「適応症X」の上市承認付与日:2027年8月1日
(データ保護期間) 上記<例1>に同様。
- N期間(遅れた年月)=1年
- 保護期間=6年-1年
=5年 - 従って、保護期間の満了日=中国での上市承認付与日から5年
=2032年8月1日
<例4>中国開発が遅れた場合(日本、中国の適応症が異なる)
(前提)
- 「創新薬A」の「適応症X」について日本での販売承認日:2025年8月1日
- 中国で「適応症Y」(日本と異なる)の上市承認申請日:2026年8月1日
(但し、中国申請の時点で、日本で「適応症Y」について未承認) - 中国での「適応症Y」の上市承認付与日:2027年8月1日
(データ保護期間)
- N期間(遅れた年月)=1年 しかし、「適応症Y」については、遅れていない。
⇒「創新薬A」(「適応症Y」)は中国では1類の創新薬として、6年間の保護期間が付与される。 - 保護期間=6年
- 従って、保護期間の満了日=中国での上市承認付与日から6年
=2033年8月1日
3.オリジナル薬のジェネリック承認(規則案§7)
日本にない制度として、海外で既に販売承認が下りているオリジナル新薬について、当該オリジナル薬が中国で未承認の場合、中国企業等の第三者(「第三者」)が中国で当該オリジナル新薬に対するジェネリック薬を申請し、販売承認が下りた場合、3年間の保護期間が付与されます。かかるオリジナル薬を海外で開発に参画している企業から中国での開発・販売権(ライセンス)許諾を受けていない、全く関係のない「第三者」が中国で開発して承認取得した場合の話です。
当該オリジナル薬が海外で上市承認が下りている事実をベースに、当該第三者は、中国で確認試験(小規模のPh III)を実施すれば、中国で上市販売承認を得られます。尚、医薬品登録分類上は、「3類」として分類されます。
これは、一種のジェネリックの扱いですが、「第三者」は、中国で小規模ながら臨床試験を実施して臨床データが生まれることから、かかるデータに対して、3年間、データ保護が付与されます。従って、3年間は、それ以外のジェネリック企業は当該データに依拠してBE試験をしてジェネリック薬として申請しても、上市承認はおりません。
もっとも、当該オリジナル薬を海外で開発している企業等は、たとえ、3年の保護期間であっても、自身が中国で当該確認試験を実施すればNMPAに申請して上市承認を取得することは可能です(規則案§3)。
4.「データ保護」制度の今後
中国当局は、前述の新薬「データ保護」規則(案)と同時にその手続規則(药品试验数据保护工作程序: https://www.nmpa.gov.cn/xxgk/zhqyj/zhqyjyp/20250319181537196.html)を公表して、一般に広くパブリックコメントを求めています。コメントの提出期限は2025年5月18日です。 ただ、この公表に先立って、主要な新薬企業やジェネリック企業等には事前の意見聴取を済ませています。従って、今後、多少の微調整はあるものの、大筋、今回の案に沿って、最終の規則が公表され、年内(今秋)には施行される予定とされています。
付録
中国政府のウェブサイトは日本からアクセスできない場合がありますので、こちらからもダウンロードできるようにしました。